


推荐产品



公司新闻/正文
惠研生物助力微生物宏基因组研究!
人阅读 发布时间:2022-04-25 13:59
采用MiSeq高通量测序仪,准确高效产出基因组数据
随着读长增加,结合pair-end测序及拼接技术的使用,Miseq测序仪已经成为取代454测序的理想工具。
惠研生物(BioGenius)竭诚为您提供16S rDNA Miseq测序分析服务。
微生物16S rDNA 测序服务
16S rRNA基因,即16S rDNA,长度约1.5Kb, 含有高度保守的基因片段,同时不同菌株之间也含有变异的核酸片段。因此16S rDNA可以体现不同菌株之间的差异,称为理想的鉴定菌株的目标序列。通过对其序列的测序分析,可以判定不同菌种,菌属间遗传关系的远近。随着读长增加,结合pair-end测序及拼接技术的使用,Miseq测序仪已经成为取代454测序的理想工具。惠研生物(BioGenius)竭诚为您提供16S rDNA Miseq测序分析服务。
在细菌的系统分类学研究中最常用的分子钟是rRNA,其种类少含量大,分子大小适中,存在于所有物种中,特别是其进化具有良好的时钟特性,在结构与功能上具有高度的保守性。rRNA是细菌乃至真核细胞核糖体的基本组成部分,编码rRNA的基因按5’-16S-23S -5S-3’顺序方式排列,由两个非编码的间隔区域所分开。16S rDNA, 23S rDNA, 5S rDNA三个部分组成一个RNA操纵子,这个操纵子作为一个单位进行转录。转录后处理成的成熟16S ,23S 和5S rRNA,其结构既具有保守性又具有高变性。保守性可以反映出生物物种的亲缘关系,高变性则揭示物种的特有特征基因组核酸结构序列,是鉴定物种的分子基础。
应用16S rRNA基因来确定原核生物的亲缘关系已成为原核生物系统发育和分子生态学研究中公认的经典标准。然而,该基因在原核生物内往往同时存在多个拷贝,而且拷贝之间的基因序列并不完全一致。因此,在原核生物生态学研究中,基于16S rRNA基因的菌群多样性分析会引起一定程度的高估。
目前,有研究报道从2013个测序的原核生物基因组中,对16S rRNA基因的拷贝数及基因组内部异质性做了详细的分析研究,发现:对于经常用来进行焦磷酸测序的区域中,V4-V5区域显示了最低的高估程度(约为3.0%),而V6区域的高估程度最高(约为13%)。因此在原核生物分子生态学研究中,16S rRNA基因的V4-V5区域更适合作为测序的目的片段。(Applied and Environmental Microbiology,19 July 2013;DOI:10.1128/AEM.01282-13)
惠研生物的优势
- 拥有标准化操作实验室和高通量测序技术平台,实验周期短,质量可靠。
- 拥有Illumina HiSeq 2500、MiSeq等多种高通量测序平台。
- 技术人员经验丰富,可以根据合作伙伴要求提供实验方案、解决实验问题、分析实验结果。
- 拥有专业的生物信息团队和超算服务器,可为合作伙伴提供全面生物信息分析服务与技术支持。
基础数据分析模块:
1. 原始数据整理、过滤及质量评估
1.1 原始数据处理与样本序列数目统计
1.2 序列长度分布
2. OTU列表生成及注释
2.1 OTU 计算
22 OTU heatmap热图
3. 物种丰度分析
3.1 稀释曲线分析
3.2 Alpha多样性分析
3.3 物种丰度差异分析
3.4 差异物种丰度聚类分析
高级数据分析模块:
4. 群落结构分析
4.1 单样品物种分布(按门、属整理)
4.2 多样品物种分布
4.3 进化关系分布
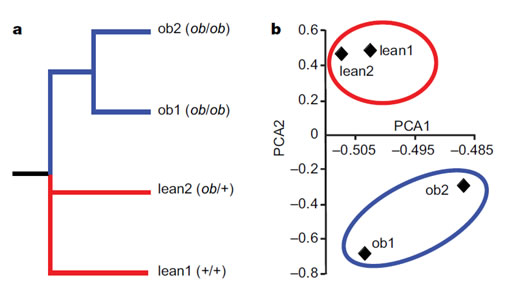
4.4 Beta多样性分析 (PCA分析)
16S rDNA 综合分析:群落多样性分析
样本需进行16s rDNA测序(MiSeq/454/Sanger;ABI;3730xl),尤其对16s;RNA的V2/v3等可变区和(或)V6;超可变区进行深度测序(amplicons测序)。
- 本方案对16s测序序列进行操作分类单位的计算(operational;taxonomic;units,OTUs),依据16s rRNA;Greengene数据库比对的最佳匹配结果赋予样本分类标签(Taxonomy)。
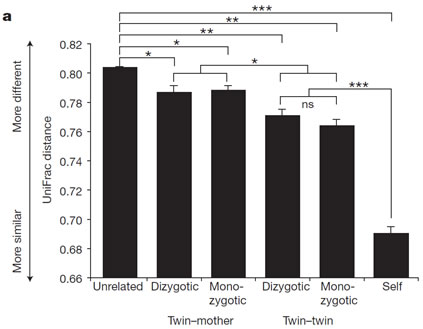
- 构建邻接树,进行UniFrac聚类分析与PCA分析:利用每OUT代表性序列构建邻接树;并通过邻接树计算UniFrac距离。依据UniFrac距离进行主成性分析。系统发生聚类分析采用UniFrac方法,基于的原则是群落可以依据他们共同的进化历史进行比较,用他们共享系统发育树上的分支长度的程度来衡量并可采用t-test检验统计组间UniFrac距离上是否存在种群显著性差异。除UniFrac之外,也可基于分类(taxon-based)的方法分析。
- Rarefaction;与系统发生多样性测度(phylogenetic;diversity;measurements)计算,分析样本中种群多态性程度。;构建样本的系统发生多样性累计曲线(Phylogenetic;diversity;curves)评估样本之间的群落差异性。


16S rDNA综合分析:群落生物功能KEGG通路分析
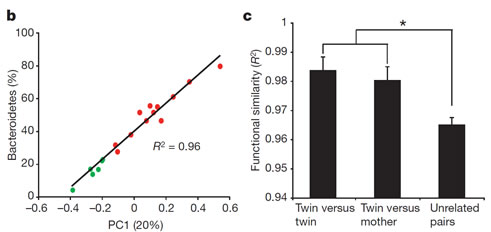
本方案对16sRNA样本群落和基因功能进行系统学分析。
- 通过与KEGGPathway通路与STRING数据库的BLAST比对,以及与项目相关的个性化选定的相关数据库(如人类肠道微生物数据,环境微生物基因组数据库,CAZymes数据库(carbohydrate-activeenzymes))比对,综合分析获得基因功能群体聚类。
- 依据样本的KEGG通路富集度进行层次聚类
- KEGG通路富集度PCA-主成性分析,并对第一成分种群与其相对丰度进行相关性计算;
- KEGG通路富集度样本组别之间的比较(采用t检验)。
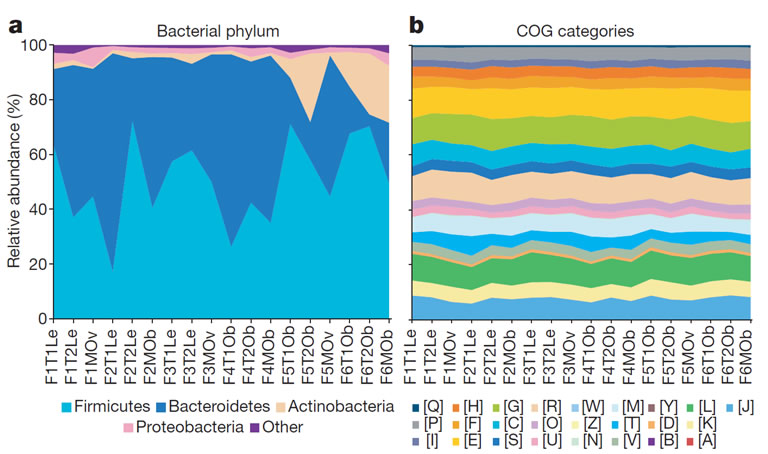
16S rDNA综合分析:群落生物功能COG通路分析
本方案对样本宏基因组16srRNA测序数据分析获取样本的主要门类和基因功能:
(1)进行NR数据库比对确定主要门类以及在样本内各自相对丰度;
(2)通过与COG数据库比对分析,获得各自样本内关于基因功能相对富集度(百分比)。

第三代单分子测序基因组完成图,火热促销中!
更多促销信息,试试关注“惠研生物”官方微信平台,发送关键词“促销”查询吧。
惠研生物开创科研新思路,欢迎来电咨询。
惠研官网:www.biogenius.cn
咨询热线:400-016-9606/180-1908-2932
咨询邮箱:service@biogenius.cn
询价列表

暂时没有已询价产品
